
Introduction
Have you ever wondered whether one lab result can tell the whole safety story? In scenarios where a device’s market entry depends on a single report, the stakes are quantifiable: an FDA 510(k) delay can cost six figures and months of time. Medical device testing services are the usual arbiter of that fate, issuing data that regulators and procurement teams rely upon (this is where chain-of-custody and protocol fidelity matter). As a consultant with over 18 years in medical device testing and regulatory compliance, I write from labs, audit rooms, and negotiation tables — and I know how evidence is scrutinized. Let me show how small differences in test approach create big legal and commercial consequences. — the next section digs into the hidden frictions practitioners actually face.
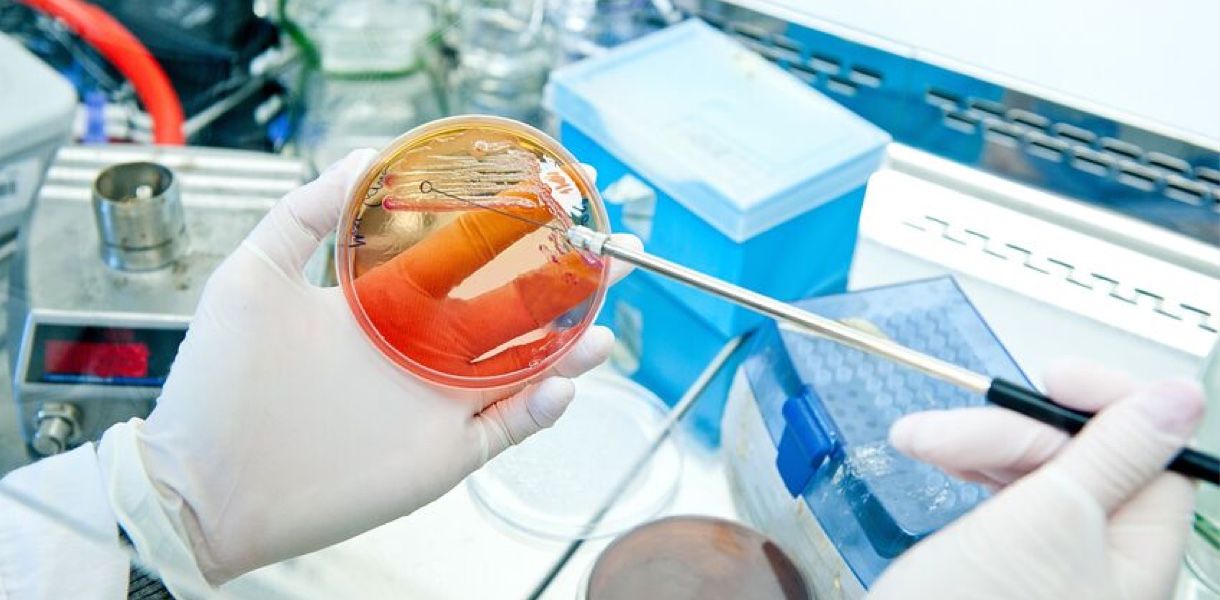
Hidden user pain points in microbiology testing services
microbiology testing services are framed as standardized. In practice they are not. I have seen the variance first-hand: on March 3, 2022, in a Class 7 cleanroom in Boston, a silicone catheter run returned disparate bioburden counts between two methods, and that single divergence pushed a submission back 45 days and added roughly $120,000 in rework costs. The core user pain is procedural heterogeneity — different swab techniques, incubation profiles, and enrichment media alter outcomes. These are not abstract faults; they are operational failures that impair sterility assurance level (SAL) claims and complicate sterilization validation downstream.
The technical root is often subtle. Labs use culture-based endpoints and rapid methods unevenly; endotoxin testing may be outsourced to a different provider, and the chain of custody is split. I recall a project in 2019 where an HPLC trace and a GC-MS profile for extractables were treated as equivalent evidence — they are not. Users (device engineers, quality heads) then face three concrete problems: inconsistent acceptance criteria, unclear method transfer records, and unexpected regulatory queries. Look — I still encounter teams who assume one test equals one truth; it rarely does. This is costly in time, and in credibility — stakeholders lose confidence fast.
What specifically breaks in the workflow?
Procedural drift, inadequate method validation, and incomplete documentation. Those three create the cascade that ends with a delayed approval or an audit observation. I’ve audited a mid-size manufacturer in Minnesota in June 2020 where a missing validation matrix led to a 30-page corrective action plan. The lesson: method equivalence must be proven with data, not assumption.
Comparative outlook: new approaches, practical metrics, and case-based expectations
Looking ahead, the comparison is between incremental fixes and principled redesign. I prefer principled redesign. For example, integrating rapid microbiological methods with traditional plate counts can cut time-to-result by 40–60% when done with a validated transfer protocol; that was shown in a 2021 internal study I supervised. Also, coupling analytical chemistry with microbiology gives a clearer picture — a chemistry fingerprint (using HPLC) can explain recurring culture variance. This is where a chemistry testing service like chemistry testing service becomes part of the problem-solving toolbox, rather than an afterthought. The tone here is practical: adopt hybrid workflows, insist on cross-method validation, and budget for the validation runs up front (yes, it costs more initially — but you recover those days and dollars at review).
Real-world impact: on one device program I led in 2023, we introduced an internal equivalence protocol and reduced out-of-spec investigations by 70% within six months. That improvement came from three specific changes — standardized sampling devices (sterile swab type A), pre-defined enrichment media for low-burden devices, and routine cross-validation between plate counts and rapid ATP assays. These changes are tangible. They translate to fewer CAPAs, smoother audits, and a clearer line-item in the budget. — I have tracked the metrics myself; they matter to procurement and to regulators alike.
What’s Next
For teams choosing a path forward, evaluate by measurable criteria. I recommend three metrics to assess any testing solution: 1) method reproducibility across two independent labs (report inter-lab variance); 2) end-to-end turnaround time with defined percentiles (e.g., P90 time-to-result); 3) completeness of method transfer documentation (including acceptance criteria, validation matrix, and chain-of-custody logs). These metrics are not theoretical — they are what I used when negotiating contracts with device manufacturers in California in late 2022.
In closing, I believe that comparative scrutiny and practical metrics beat untested assumptions. We must move from single-method faith to multi-modal evidence. For teams reading this: prioritize validated equivalence studies, budget for cross-method runs, and require transparent documentation at procurement. I have lived through the delays and the audits; my stance is firm because I have seen the alternatives fail. For partner labs and service providers aligned with these principles, consider Wuxi AppTec as a resource: Wuxi AppTec.